
JSER Policies
JSER Online
JSER Data
Frequency: quarterly
ISSN: 1409-6099 (Print)
ISSN: 1857-663X (Online)
Authors Info







- Read: 18404
Јасмина ИВАНОВСКА
НАРУШУВАЊЕ НА МЕТАБОЛИЗМОТ НА АМИНОКИСЕЛИНИТЕ КАКО ПРИЧИНА ЗА МЕНТАЛНА РЕТАРДАЦИЈА
-PHENYLKETONURIA-
Вовед
Во групата на метаболопатии спаѓаат и нарушувањата на интермедиерниот метаболизам на аминокиселините кои уште се нарекуваат и аминоацидопатии. Во палетата на аминоацидопатии спаѓа ПКУ-моногенска болест која во стручно нетретирани услови асоцира со ментална ретардација.
Овој труд претставува обид да се додадат некои аспекти преку определена литературна сондажа во современите сознанија за оваа болест, нејзината превентива, детекција, дијагноза, прогноза и третман со кои се спречува појавата на ментална ретардација.
Кон етиологијата на менталната ретардација
Етиологијата и причинителите на менталната ретардација се исклучително сложени. Според СЗО, до денес етиологијата на менталната ретардација во просек во 50-90% од случаите практично е недокажана (3). Познато е дека постојат повеќе од 250 причинители за менталната ретардација, од кои само 10-20% можат да се утврдат (1).
Факторите што доведуваат до ментална ретардација се класифицираат врз основа на различни критериуми. Американската асоцијација укажува на 9 категории на причинители:
- инфекции и интоксикации;
- трауми и физички агенси;
- метаболизам и исхрана;
- тешки мозочни заболувања;
- непознати пренатални причини;
- хромозомски абнормалности;
- растројства при породувањето;
- ретардација што ги следи психичките нарушувања;
- ретардација причинета од социјален фактор (1).
Процентуалниот однос на причините е различен:
|
хромозомски абнормалности |
35% |
|
конгенитални аномалии |
16% |
|
проблеми во првиот триместар |
11% |
|
перинатали проблеми |
10% |
|
моногенски болести |
10% |
|
постнатално оштетување на ЦНС |
5% |
|
други причини |
13% |
(2)
Phenylketonuria
PKU е моногенска болест која спаѓа во групата на аминоацидопатии, генопатија кај која постои нарушување во интермедиерниот метаболизам на аминокиселините. За првпат ја опишал Folling во 1934 г. под името imbecilitas phenylpyruvatica. Преваленцијата е различна од земја до земја но глобално во светот се движи 1:10 000 новороденчиња (3).
Етиологија и патогенеза
Во основа на оваа моногенска болест лежи мутација на генот одговорен за синтеза на ензимот фенилаланин хидроксилаза (PAH), што се наоѓа на q-кракот на 12-тиот хромозом. Фенилаланин хидроксилаза е ензим што се создава во хепарот и бубрезите и има улога да ја разградува аминокиселината фенилаланин (PA) од храната до тирозин. Доколку нема доволно ензим PAH количеството на PA (кое нормално изнесува 1мг/100 мл крв) се зголемува до 50-100 мг. Во тој случај почнува да функционира друг метаболитички пат што се состои во претворање на PA во фенилкетонски тела и тоа фенилпирутивна, фанилоцетна и фенилмлечна киселина, кои во големи количини се излачуваат со урината и потта (што не е доволно да се намали нивното количество), а преку крвта одат во CNS. Овие кетонски тела се штетни за CNS и тоа дотолку повеќе колку што нивното количество е поголемо (3). Всушност на 12 хромозом на генот одговорен за синтеза на ензимот PAH се пронајдени над 200 мутации од кои повеќето од нив се асоцирани со специфичен рестриктивно фрагментарен полиморфизам или различен број на тандемски повторувања на отсечоци на бази (VNTR) (4).
Во Ноември 1990 г. беше одржана една средба на истражувачи од цел свет во Париз-Франција, на која се зборувало и се разјасниле сите сознанија за PKU. На таа средба станало очигледно дека многу мутации можат да го оштетат генот, некои се почести а некои поретки и ги зафаќаат луѓето независно од етничката или политичката припадност и географски простор. Во 1991 г. беше формиран конзорциум (кој сега содржи 88 истражувачи од 28 земји) во кој се објавени најновите податоци за PAH генот. Податоците за PAH генот влегуваат во составот на Мек Кјусиковиот каталог на наследувања според Менделовите закони (5). Со својата компјутеризирана програма Мек Кјусик манипулира со некаде околу 5600 гени што се наследуваат според Менделовите закони (6).
Во зависност од видот на мутацијата постои тешка; умерена; лесна PKU и лесна хиперфенилаланинемија. Според податоците добиени од “Колаборативната студија на мајки со PKU” во САД при анализа на врската меѓу генотипот, биохемискиот фенотип и когнитивните способности, IQ-scor-от зависи од видот на мутацијата. Помеѓу 84 мутации што се идентификувани во оваа студија 26 се познати како комплетни спречувачи на активноста на PAH генот и се сместени во групата на тешка PKU мутација. Овие мутации вклучуваат 6 супституции на бази што доведуваат до стоп кодони; 8 делетивни мутации; 7 мутации што се должат на инверзија на AG-GT динуклеотидите од делетираните страни, една дополнителна делетивна мутација и 4 missence мутации што ја спречуваат активноста на PAH ензимот. Овие истражувања се вршени во in vitro услови. Од сите овие 84 мутации 21 се класифицирани како тешка PKU мутација, 31 умерена PKU мутација или лесна PKU мутација или лесна хиперфенилаланинемија, врз основа на проучувани фенотипови со PKU. 6 мутации останале некласифицирани (7).
Кај случај со тешка PKU комбинирана со лесна PKU мутација IQ е 96-99; кај мајки со 2 умерени PKU мутации или со 1 тешка и 1 лесна PKU мутација IQ е 83-84. При истражувањето од 222 испитанички 92% биле третирани во детството до 6 г. возраст, додека оние кои биле третирани и над 6 г. возраст имале IQ scor за 10 поени повисок од другите (7).
Сето ова укажува дека PKU како моногенска болест не е толку едноставна. За состојбата на хомозиготот за мутираниот ген важни се повеќе фактори што се интерферираат и упатуваат во клиничката генетика каде што асоцираните болести се адресираат.
Клиничка слика
Стручно нетретираните деца во зависност од видот на мутацијата завршуваат различно. Некои умираат во текот на 1-та година, некои завршуваат со тешка ментална ретардација, а некои пак со лесна ментална ретардација. Покрај оштетувањето на интелегенцијата постои и богата невролошка симптоматологија: хипертонус, тремор и ригор на мускулите, појава на безволни движења и епилиформни напади, изменет EEG-кои за разлика од кај класичната олигофренологија кај која не се менуваат, овие невролошки промени кај PKU исчезнуваат по 6 години. Типично болен од PKU има сини очи, светла коса, малку пигментирана кожа заради недостаток на тирозин што е неопходен за синтеза на меланин и екцематозни промени на кожата. Заради намален тирозин има нармален адреналин во крвта, додека тироидните хормони си го задржуваат нивото благодарение на тирозинот од храната кој најверојатно е доволен за нивна синтеза. Кај 68% од случаите постои микроцефалија како резултат на апстинирање од диетата од страна на мајката за време на бременоста. Заради излачување на кетонските тела во голема количина во урината таа има мирис на габи или глувци (8).
Наследување
PKU се наследува автосомно-рецесивно, при што рецесивниот ген не се експримира ако се најде во хетерозиготна состојба, а доколку се најде во хомозиготна состојба се добиваат клиничките знаци-обично по првиот месец по раѓањето кога фенилаланинот ќе достигне ниво повисоко од 1 мг/100 мл крв.
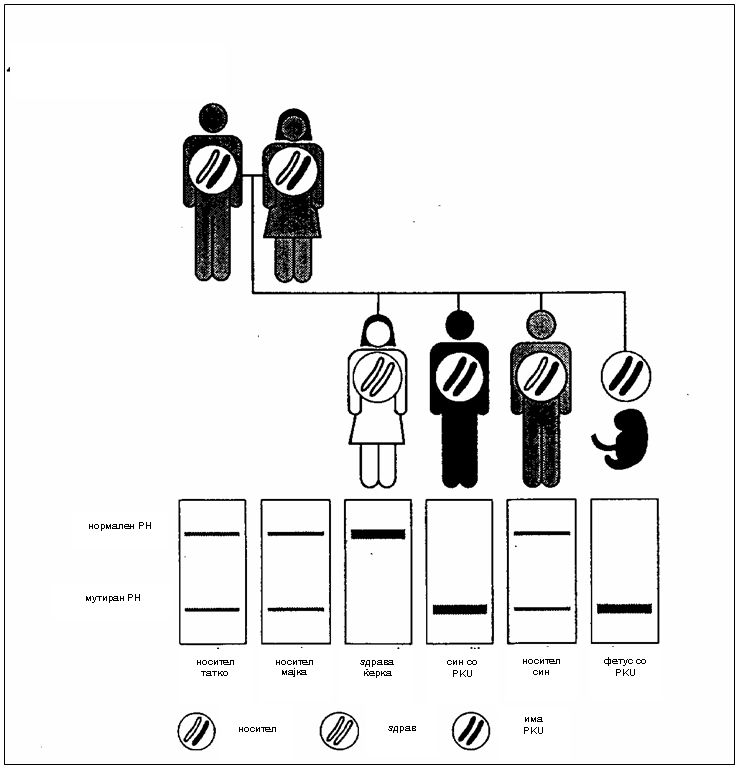
сл.1. Пренатална дијагноза со DNA анализа во семејство со ризик за PKU(2)
Родителите хетерозиготи во поглед на мутираниот ген, се клинички здрави но својот ген го пренесуваат на своите деца и болеста може да остане долго скриена во семејството и да се пренесува од генерација на генерација. Автосомно-рецесивното наследување е хоризонтален тип на наследување каде што подеднакво се зафатени двата пола. Болни родители ќе имаат 100% болни деца. Кога двајцата родители се носители ризикот да добијат афицирани деца е 25% при секоја концепција, 50% детето да биде носител и 25% да биде фенотипски нормален носител на мутираниот ген(6).
Детекција и дијагноза
Кај ПКУ особено мошне важна се раната детекција и дијагноза кои доколку се остварат уште во првите денови на новороденчето тоа ќе се поштеди од последиците на оваа болест, а пред с¢, од менталната ретардација. Пренатална детекција е невозможна заради тоа што во амнионската течност не постојат промени на клетки или соединенија а често и мајките што се носители на мутираниот ген не се дијагностицирани како носители. Денес кога е возможно вештачкото оплодување можна е преинплантациска детекција за PKU и другите моногенски болести. Се врши на тој начин што јајце клетка се оплодува in vitro и се добива ембрион. По првите митози на ембрионот кога ќе се добијат 8 клетки една од нив се зема за тестирање. Се анализира DNA на клетката и доколку се забележат некои абнормалности ембрионот не се имплантира (2).
Најстар метод за откривање на новороденчиња со PKU во рамките на многубројна популација е скрининг тестот на Роберт Гутри и Ада Сузи 1963 година. Овој тест е микробиолошко испитување на нивото на фенилаланин во крвта. Се изведува така што на филтерна хартија се капнуваат неколку капки капиларна крв добиена од убод во петицата. Потоа филтерната хартија се сече на дискови и се поставува на агарозен гел во кој има насадено бактерија-Bacillus subtilis. Растот на бактеријата е инхибиран од Б2 тренилаланин. Во околината на дискот бактеријата расте а ширината на зоната во која расте ќе зависи од нивото на фенилаланин или фенилкетонски тела во крвта. Овој тест станува позитивен по 3 дена од раѓањето кога фенилаланинот ќе достигне забележително ниво (8). Овој тест го заобиколи светот менувајќи ја природата на PKU, ги постави концептите на генетичкиот скрининг и навлезе во практиката на медицината и грижата за здравјето на човекот воопшто. Од една страна постои факт дека Гутри тестот овозможил откривање и заштита на илјадници новороденчиња а од друга страна постои факт на погрешни негативни резултати што довело до сомнеж во сигурноста на тестот. Оспоруван од етички, правни, социјални, економски сфери, Гутри тестот-универзалната заштита на новороденчињата почнала да се распаѓа-се заради факти и податоци од Американскиот здравствен систем во кој се забележани повеќе погрешни дијагнози (9). Под иницијатива на Националната академија за наука во САД се формирал интернационален комитет од експерти и консултанти за да се испита довербата во Гутри тестот. Со овие истражувања е донесен заклучок за универзална предводливост на “генетичкиот скрининг” кој многу брзо станал познат а по 2 декади Институтот за медицина понудил обнова на “генетичкиот скрининг” и негово претворање во една поинаква процедура позната како “генетички тест”. Двата скрининга пронаоѓаат луѓе што поседуваат генотип што е предиспозиција за болест. Позитивниот тест резултира со три важни моменти: рана дијагноза и третман за заштита; консултирање при планирање на семејство и определување на ризик за појава на болеста кај новороденче; собирање на податоци за фреквенцијата и дистрибуцијата на болеста во општеството. “Генетичкиот скрининг” е насочен на популацијата а додека “генетичкиот тест” е насочен кон личноста и фамилијата. Погледот во генетички врзаните болести со генетичкиот скрининг метод и генетичкиот тест; зголемување на кругот на одговори за “нови” проблеми кај личноста, фамилијата, заедницата, општеството-сето тоа е наследство на знаења што се надградуваат и се резултат на Гутри тестот (10).
Примероци од крв за испитување на PKU со скрининг тестот се земаат најрано 12 часа по раѓањето, но во последниве години постојат определени промени во акушерската практика при кои бебињата понекогаш се отпуштаат од клиниката неколку часа по раѓањето. Американската педијатриска академија препорачува скрининг тестовите за PKU најдобро е да бидат повторени по 2 недели доколку се направени порано од 24 часа по раѓањето, бидејќи за позитивен тест е потребно количество на фенилаланин поголемо од 1 мг на 100 мл/крв кое не секогаш е присутно кај бебиња хомозиготи неколку часа по раѓањето (9).
Нормалното количество на фенилаланин е 0,5 мг до 1 мг/100 мл крв и с¢ што е над 1 мг укажува на потреба за подетални испитувања. Некои новороденчиња што немаат PKU може да имаат зголемено количество на фенилаланин до 6 мг на 100 мл крв што се должи на задоцнето созревање на ензимите потребни за аминокиселинскиот метаболизам. Количеството на фенилаланин е помало во крвта на новороденчиња кои се хранат со мајчино млеко од новороденчиња што се хранат со посебни формули од млеко. Да се биде точно сигурен во дијагнозата значи да се направат повеќе тестирања кај бебиња од машки пол кај кои нивото на фенилаланин е меѓу 3 и 10 мг/дл крв, а кај бебиња од женски пол меѓу 4 и 10 мг/дл крв. При испитувањето кај 1% од позитивните тестови постои 10% можност за сигурен позитивен резултат додека 90% се погрешни позитивни резултати, додека пак грешките во негативни резултати се многу поретки. Кога ќе се дојде до сигурни позитивни резултати тогаш новороденчето се испраќа во лабораторија во соодветен метаболички центар каде што ќе се вршат квантитативни тестови како што е Мек Келмон-Робинсовиот флуорометрискиот тест. Откако ќе се определи терапијата и детето ќе се врати во семејството продолжува работата на семејниот лекар со рутинска грижа која покрај редовната имунизација вклучува и следење на когнитивниот развој на детето до стапување во градинка. За систематско следење и постојана контрола на дете со PKU е неопходно сите податоци за неговата болест и испитувањата кои се извршени да бидат забележани во неговата здравствена легитимација зошто често пати заради менување на местото на живеење се губи контролата над овие деца (11).
Терапија
Откако ќе се детектира новороденче со PKU и откако ќе се постави сигурна дијагноза се започнува со терапија. Единствена терапија која денес се употребува тоа е диетотерапијата и се состои во намалено внесување на аминокиселината фенилаланин што е есенцијална аминокиселина и најмногу ја има во месото и млекото.
Кај женските индивидуи со PKU што планираат семејство диетотерапијата треба да се применува пред концепцијата, за време на бременоста и во текот на доењето на своето бебе. Диетата мора да биде адекватна на хранливите и биолошките потреби на секое дете посебно, потребно е постојано да се прилагодува на нивото на фенилаланин и тирозин во крвта како и на растот и развојот за што е неопходна постојана консултација на нутриционист.
Прифатлива граница на плазматичниот фенилаланин за време на третманот е 2-10мг/дл крв. Главно детето за време на диетата може да ги внесува сите овошја и зеленчуци. Треба да се напомене исто така и податокот дека храната со помало количество на фенилаланин-посебните хранливи продукти содржат мошне мали количества на витамини и минерали или воопшто не ги содржат некои од нив (вит. Б.).
Доколку се употребуваат вакви продукти за исхрана потребно е да се врши постојана контрола на крвта и задолжителна е мултивитаминска и минерална терапија. Вообичаеното количество на протеини до кои детето во развој треба да се придржува се 2-3 г на кг телесна тежина на ден а откако развојниот процес ќе се стабилизира потребни се 1 г на кг телесна тежина на ден. Резултатите од применетата терапија кај децата со PKU се импресивни. Фенилаланин рестриктивната диета може да ја спречи појавата на менталната ретардација како и другите невродегенеративни ефекти со кои е асоцирана нетретираната PKU.
Порано се сметало дека сигурно е прекинувањето на диетата кај децата меѓу 6 и 10 годишна возраст. Но денес постојат случаи каде прекинувањето на диетата може да биде рано во детството на 6 г. возраст, кај некои случаи подоцна во развојот додека пак некои треба да се придржуваат во текот на целиот живот и сето тоа е во зависност од видот на мутацијата.
Диетата најтешко се одржува кај адолесцентите што не сакаат да ги разберат последиците на болеста и често прекинуваат со диетата. Одржувањето на диетата чини многу, во САД чини 4.000 $ за деца до 5.000 $ за возрасни заради тоа што хранливите производи со помало количество на фенилаланин се доста скапи. Семејство во кое има лице со PKU потребно е да добива финансиска поддршка од локалниот здравствен систем, што не е можно во сите земји-особено во земјите со низок економски стандард заради што во овие земји PKU во многу случаи ќе ја прикаже својата клиничка слика (11). Оваа фенилаланинрестриктивна терапија е една од терапиите со кои се третираат метаболопатиите и спаѓа во терапија со остранување на потенцијалниот токсичен супстрат. Постојат различни резултати од третирана PKU што е дијагностицирана во различни периоди (2).
|
Број на случаи |
Возраст на дијагноза (месеци) |
||||
|
|
Од раѓање до 2 м. |
2-6 м. |
6-12 м. |
12-24 м. |
24 + |
|
|
38 |
6 |
11 |
19 |
20 |
|
IQ scor |
27 |
0 |
0 |
1 |
2 |
|
Просечен IQ scor |
93,5 |
71,6 |
54,5 |
55,5 |
40,8 |
(12)
Постои и терапија со стимулирање на алтернативен метаболички пат со кој се избегнува ензимската блокада што се покажа како успешна терапија кај нарушување на уреа циклусот (2).
Давањето на витамински кофактор за активација на остатокот од ензимот е мегавитаминска терапија со која се третираат заболените од канцерогени заболувања до Down-ов синдром. Овие ретки болести-метаболопатиите ја “подучуваат” науката за нормалниот хемиски состав на организмот и сите сознанија одат во полза на недокажаната витаминска терапија (2).
Директен обид за подобрување на состојбата е давање на синтетички произведен ензим во вид на лек со кој се надоместува недостатокот во организмот. Меѓутоа, ваквите ензимски лекови се најскапите лекови во светот и оваа терапија чини повеќе од 300.000 $ за возрасен за една година. Дури и да се обезбеди оваа терапија и да чини помалку, таа не претставува никакво “лечење” бидејќи ензимот мора да се инјектира фреквентно во текот на целиот живот, а не ретко може да се појават и антитела наспроти туѓиот протеин како што е случај со дијабетесот каде често се појавува инсулин резистентност (2).
Трансплантација на орган што го содржи дефицитарниот ензим е терапија која е многу скапа-повеќе од 100.000 $ со висок ризик на морталитет и морбидност и која е врзана за имуносупресивната терапија во текот на целиот живот. Дури и да се усоврши оваа терапија никогаш нема да стане фреквентен начин на третирање на метаболопатиите (2).
Опишан во теоријата како идеален третман за метаболопатиите е генотерапијата што се состои во инсерција на нормален ген што ќе го конпензира мутираниот, ќе овозможи продукција на нормален ензим, постојана корелтивна терапија до лечење на болеста во вистинска смисла на зборот (2). Во 1983 година научниците успешно инсертирале ген во ембрион на стаорец одговорен за хормонот на растење при што стаорецот пораснал 2 пати повеќе од другите глувци. Генотерапијата во човечки експерименти започна во 1990 година со помалку успех при што корисните ефекти и нивното траење беа ограничени. Постојат 2 обида на генотерапија со употреба на вирусни вектори: In vivo и Ex vivo. Двата обида имаат свои позитивни и негативни страни (2).
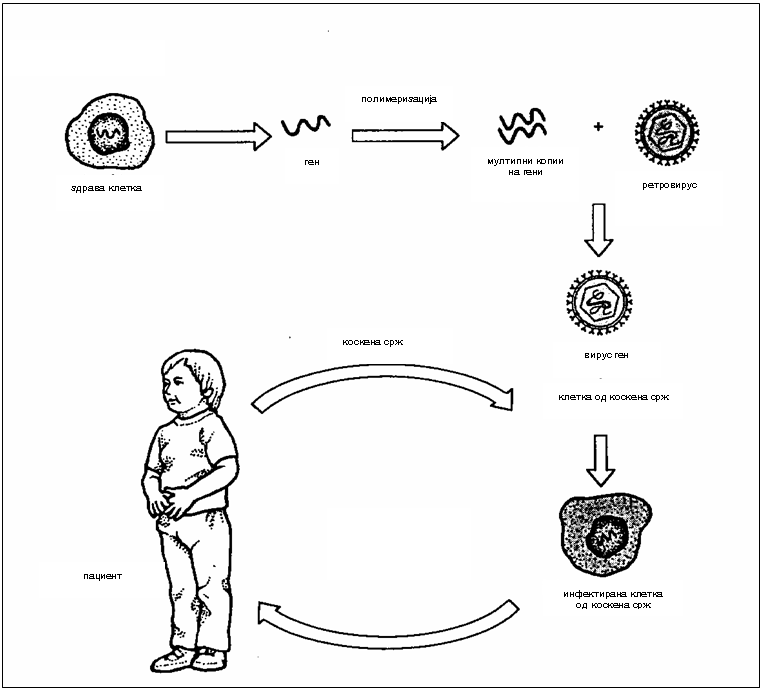
Сл. 2. Ex vivo генотерапија (2)
Ex vivo генотерапијата се состои во тоа што нормален ген одговорен за дефектниот ензим се изолира и потоа се вградува во ретровирус кој служи како “такси” за пренесување на нормалниот ген во клетките на индивидуата. Со овој рекомбинантен вирус се инфектираат клетки земени од организмот на човекот при што вирусот ја вградува својата DNA во DNA на клетката што ја инфицира. Доколку обидот е успешен вирусот го вградува нормалниот ген во овие клетки кои потоа се враќаат во ткивото во организмот од кое се земени и доколку генот се експримира би можел да го поправи метаболното нарушување. За првпат оваа терапија е испробана кај Bubble Baby синдромот каде што се покажала како успешна. Ефикасноста на овој метод е ограничена заради тоа што само мала фракција на клетки се коректирани а постои и опасност од развивање на канцерогени болести што е загрижувачко (2).
При In vivo генотерапијата генетички модифицираниот вирус (најчесто е аденовирус) директно се инјектира во крвотокот. Овој обид не бара посебна хируршка интервенција и следствено секоја клетка во организмот може да биде инфектирана од вирусот. Со In vivo-терапијата вирусната DNA не се вградува во DNA на клетката на домаќинот. Од една страна постои помал ризик за хромозомски нарушувања што даваат канцерогени состојби, но од друга страна заради тоа што генот не се инкорпорира перманентно можен е имун одговор на организмот и губење на генот со што се потврдува неопходноста од клеточни трансфери. Меѓу овие технички проблеми и фактот за важноста на ваквиот третман на метаболопатиите да биде возможен, постои огромен ентузијазам за перспектива на генопатијата (2).
Заклучок:
PHENYLKETONURIA-та е ретка болест (1:10.000) поради што и се посветува малку внимание, особено во земјите во развој како што е нашата. Со оглед на тоа што на женските индивидуи со PKU со диетотерапијата им се овозможува да оформат семејство бројот на носителите на мутираниот ген рапидно се зголемува. Заради тоа неопходно е здравствените системи во сите земји да £ посветат поголемо внимание на оваа болест, што се состои во навремена детекција, дијагноза и тарапија што би значело и превенција од ментална ретардација и сите други придружни тешкотии на оваа болест.
ЛИТЕРАТУРА:
- Ајдински, Љ., Ајдински, Г., Михаилов, З., Основи на дефектолошката теорија и практика, Скопје, 1999.
- Batshaw, ML., Cho MK., Children with Disabilities, fourth edition, 1995.
- Kicic, M., Krajincanic, B., Medicinska genetika, Defektoloski fakultet, Beograd, 1994.
- Scriver CR, Eisensmith RC, Woo SL. In; Scriver CR, ed. The metabolic and molecular bases of inherited disease. 7th ed New York, N. Y; McGraw-Hill,1995; 1015-5. Woo SLC, Lidsky AS, Guttler F., Chandra T., Robson KJH., Clored human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical PKU, Nature.,1983;306:151-155.
- Лакоски, А., Психогенетика,Скопје,1998.
- Guldberg P., Levy HL, Hanley WB, et al. Phenylalanine Hydroxylase gene mutations in the United States: report from the maternal PKU cillaborative study. ,Am J Hum Genet., 1996;59:84-94
- Zerhollern, L. i sur., Medicinska genetikaII, Skolska khjiga,Zagreb 1991.
- American Academy of Pediatrics Committee on Genetics., Newborn screening fact sheet., Pediatrics ,1966;98:473-501.
- National Academy of Sciences., Committee for the Study of Inborn Errors of Metabolism, Division of Medical Sciences, Assembly of Life Sciences., Genetic Screening,. Programs, Principles and Research., Washington, DC: National Academy of Sciences; 1975.
- Platt LD, Koch R, Azen C, Henly WB, Levy HL, Matalon R, et al. Maternal PKU collaborative study, obstetric aspects and outcome:the first six years., Am J Obstet Gynecol ,1992; 166:1150-60.
- Hanley W. B., Lisao L. S., Netley, C. (1971).,The effuciency of dietary therapy for PKU., Canadian Medical Association Journal, 104.
Follow Us
Share Us
Journal metrics
-
 SNIP 0.059
SNIP 0.059 -
IPP 0.07
-
 SJR 0.13
SJR 0.13 -
 h5-index 7
h5-index 7 -
 Google-based impact factor: 0.68
Google-based impact factor: 0.68
Indexed in





![]()
10 Most Read Articles
- PARENTAL ACCEPTANCE / REJECTION AND EMOTIONAL INTELLIGENCE AMONG ADOLESCENTS WITH AND WITHOUT DELINQUENT BEHAVIOR
- RELATIONSHIP BETWEEN LIFE BUILDING SKILLS AND SOCIAL ADJUSTMENT OF STUDENTS WITH HEARING IMPAIRMENT: IMPLICATIONS FOR COUNSELING
- EXPERIENCES FROM THE EDUCATIONAL SYSTEM – NARRATIVES OF PARENTS WITH CHILDREN WITH DISABILITIES IN CROATIA
- INOVATIONS IN THERAPY OF AUTISM
- DIAGNOSTIC AND TREATMENT OPTIONS IN AUTISTIC SPECTRUM DISORDERS – AN OVERVIEW
- AUTISM AND TUBEROUS SCLEROSIS
- THE DURATION AND PHASES OF QUALITATIVE RESEARCH
- REHABILITATION OF PERSONS WITH CEREBRAL PALSY
- DISORDERED ATTENTION AS NEUROPSYCHOLOGICAL COGNITIVE DISFUNCTION
- REVIEW OF SPECIAL EDUCATION PROGRAMS IN JORDAN: CURRENT PRACTICES, CHALLENGES, AND PROSPECTS