
JSER Policies
JSER Online
JSER Data
Frequency: quarterly
ISSN: 1409-6099 (Print)
ISSN: 1857-663X (Online)
Authors Info







- Read: 18037
Владимир ТРАЈКОВСКИ
Вовед
Митохондриите се органели чија што улога е во енергетското снабдување на клетката. Нивниот број во клетките варира од неколку стотини до неколку илјади, во зависност од количеството енергија што í е потребна на клетката. Митохондриите имаат променлива големина и облик, така што некои имаат топчест облик и дијаметар само неколку стотина нанометри, додека други имаат долгнавест, стапчест изглед со должина од 7 микрометри. Внатрешноста на митохондриите е исполнета со желатинозен матрикс кој содржи големи количини растворливи ензими, потребни за добивање енергија од хранливите материи. Овие ензими ја вршат оксидацијата на хранливите материи, при што се создава CO2 и вода. Ослободената енергија се користи за синтеза на ATP, кој се ослободува од митохондриите и дифундира во клетката за да се искористи за клеточните функции (1).
Митохондријалната медицина има експлозивен растеж во последната деценија, кога повеќето од 50 митохондријални ДНК мутации се идентификувани кај афецираните пациенти. Раното детектирање и дијагностицирање на митохондријалните заболувања со помош на молекуларната генетика создава услови за брз почеток со терапија и намалување на прогресијата на заболувањата. Во наши услови многу малку се мисли на митохондријалните заболувања како причини за менталната ретардација, глувоста, слепилото или за телесната инвалидност и затоа нивното проучување е од извонредна важност за дефектолошката теорија и практика
Митохондријално наследување
Во митохондриите се наоѓа митохондријална ДНК (мт ДНК), што се нарекува “25 хромозом” (3). Таа е прстенеста, двојноверижна ДНК молекула со комплетно познати секвенци од 16.569 нуклеотиди. Сите гени во мт ДНК се добро дефинирани и тие се одговорни за кодирање на 22 транспортни РНК кои што се неопходни за митохондријална протеинска синтеза, две рибозомални РНК (12s и 16s) и 13 протеини одговорни за оксидативна фосфорилација (2).
Трансмисијата врзана за мт ДНК е од мајчинско потекло, затоа што при оплодувањето мајчинската гамета останува целосна, задржувајќи ја цитоплазмата, за разлика од сперматозоидите што ја губат. Заради ова митохондријалното наследување се вика мајчинско или цитоплазматско. Мајката е онаа што е трансмитер во ова наследување каде што обата пола се подеднакво застапени и со ист интензитет, а варијабилноста на фенотипот е честа (3,4). На слика 1 е претставено типично родословно стебло на митохондријалното наследување.
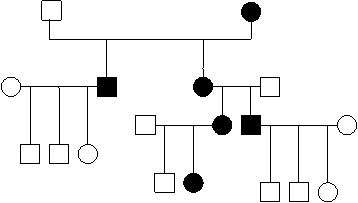
сл. 1 Типичен хередограм за митохондријално наследување |
Ефектите на митохондријалните мутации се одразуваат прво врз клетките и ткивата со високи енергетски потреби, пред сé, мозокот и мускулите, каде што се одвиваат интензивни енергетски и митохондријални оксидативни функции.
Мутациите на мт ДНК можат да водат до состојба на “хетероплазма” каде што има повеќе од еден вид на митохондријални ДНК молекули во клетката. “Хомоплазмични” клетки се оние каде што сите митохондрии имаат еднаква ДНК. Односот на хетероплазмичната и хомоплазмичната ДНК се разликува во различни ткива и може да се промени со возраста, комплицирајќи ја клиничката слика. Физиолошките ефекти на неправилната митохондријалната функција зависат од енергетските потреби на клетката. Затоа не е за изненадување што нервното и мускулното ткиво често први ги покажуваат клиничките промени. Мутациите на мт ДНК се случуваат почесто од оние во нуклеарните гени што се вклучени во оксидативната фосфорилација (2).
Многу важен факт во митохондријалната генетика е појавата на промените на мт ДНК во соматските клетки за време на стареењето. На пример, 5 килобазна делеција во митохондријалниот геном пред 40-тата година не може да даде промени на срцето. Студиите на истата делеција во мозокот ретко се манифестираат пред 75-тата година од животот, но промените се акумулираат во кортексот и базалните ганглии. Кумулативните ефекти на овие и на другите соматски мутации можат да ја променат ефикасноста на митохондријалниот метаболизам кај постарите индивидуи (2).
Класификација на митохондријалните заболувања
Митохондријалните заболувања може да се класифицираат во неколку групи. Во првата група припаѓаат оние болести што се должат на специфични мутации на митохондријалната ДНК. Тука се MELAS синдромот (myopathy, encephalomyopathy, lactic acidosis, stroke), MERRF синдромот (myoclonic epilepsy and raggrd red fibers), NARP синдромот (neurogenic muscule weakness, ataxia, and retinitis pigmentosa), Leber-овата оптичка невропатија и мала група на случаи со Leigh-ов синдром мајчински наследени. Сите овие болести се пренесуваат по мајчинска линија и обично се должат на делеции или дупликации на митохондријалната ДНК, предизвикувајќи миопатии со или без офталмоплегија каде што припаѓа Kearns Sayre-овиот синдром.
Во втората група митохондријални заболувања генетичкиот дефект се среќава во јадрените гени што ги кодираат субединиците на мт ДНК. Тука спаѓаат некои случаи на Leigh-ов синдром, Alpers-ови полидистрофии, мионеврогастроинтестиналниот синдром, Barth-овиот синдром и Friedreich-овата атаксија (5, 6, 7).
Подгрупи на некои заболувања како што се: diabetes mellitus, глувост и наследни кардиомиопатии е најдено дека се причинети од мутации на мт ДНК, а некои дефекти во мт ДНК сугерираат промени во исходот на болеста што се примарно причинети од други фактори, како што се Parkinson-овата и Alzheimer-овата болест (8).
Клинички форми на митохондријалните заболувања
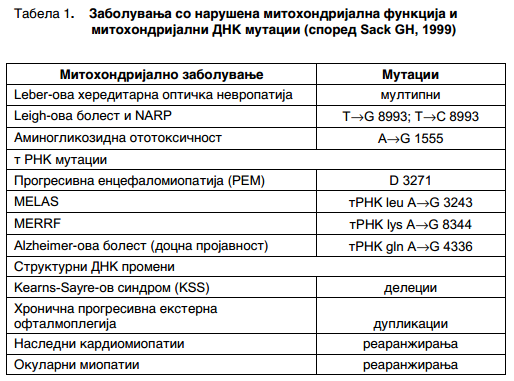
Во табела 1 може да се забележат повеќе болести причинети со митохондријални мутации.
Заболувањата асоцирани со митохондријални генски мутации се во врска со мускулната и нервната функција и често се таинствени во нивната презентација. Повеќе од овие состојби се тешко препознатливи. Тие се разликуваат врз база на наодите на мускулна биопсија и in vitro испитувањата на митохондријалната функција. Овие заболувања имаат обично задоцнета клиничка пројавност и секогаш се хетероплазматични. Различни членови во семејството (секој со еднаква мутација) може да имаат различна презентација во времето на пројавувањето на симптомите и клиничката прогресија. Возраста е веројатно важен учесник во клиничката слика и таа може да биде во асоцијација со митохондријалните ДНК промени (2).
Leigh-овата болест се карактеризира со дефицит на цитохром C оксидаза, ензим што учествува во енергетскиот метаболизам. Главните патолошки промени се во мозокот со дегенерација на сивата маса и со фокална некроза во мозокот. Во клиничката слика доминираат: хипотонија, тремор, атаксија, отсуство на тетивни рефлекси, нистагмус, стеснети зеници, слепило, хипервентилација, апнеа, диспнеа, Cheyne-Stokes-ово дишење и респираторна инсуфициенција. метаболичките нарушувања се изразуваат со нарушена глуконеогенеза, отсуство на хепатална пируват карбоксилаза и интермитентна лактатна ацидоза, на срцето се среќаваат промени од типот на хипертрофична кардиомиопатија. Во лабораторискиот наод има покачено ниво на пирувати и лактати во серумот и хипогликемија. Наследувањето е хетерогено, можно е автосомно рецесивно, Х-врзано и митохондријално. Болеста е често летална во раната детска возраст поради дегенерацијата на базалните ганглии.
Kearns-Sayre-овиот синдром: уште во 1965 година Kearns опишува девет пациенти со офталмоплегија, пигментна дегенерација на ретината и кардиомиопатија. Други значајни симптоми се слабоста на лицевата, фарингеалната мускулатура, како и мускулатурата на трупот и екстремитетите, глувоста, нискиот раст, офталмоплегијата, електроенцефалографските промени, покачувањето на протеините во ликворот. Наследувањето е митохондријално.
MERRF синдромот е карактеризиран со миоклонична епилепсија, атаксија, спастицитет, мускулна слабост со миопатија, сензоневрална глувост, искинати црвени мускулни влакна, покачени нивоа на пируватите и лактатите во серумот. Наследувањето е митохондријално и се работи за дефект во транслацијата на сите мт ДНК-кодирачки гени. Специфичната мутација на мт ДНК прв ја опишал Shoffner со сор. (1990), објаснувајќи ја како missence мутација на генот за транспортната РНК за аминокиселината лизин. A®G мутацијата на нуклеотидот 8334 се среќава кај 80-90% од случаите со MERRF. Биохемиски, мутацијата предизвикува мултипни недостатоци во ензимскиот комплекс на респираторната верига, најмногу зафаќајќи го генскиот комплекс I, односно NADH-CoQ редуктаза и цитохромот C оксидазата од генскиот комплекс IV.
MELAS синдромот (митохондријална миопатија, енцефалопатија, лактатна ацидоза, епизодни напади) клинички се карактеризира со епизодни повраќања, церебрални инсулти со хемипареза, хемианопсија, кортикално слепило и сензоневрална глувост. Наследувањето е митохондријално.
Leber-овата хередитарна оптичка невропатија (LHON) е исто така врзана за митохондријалниот интегритет. Имено, тука се работи за дегенерација на оптичкиот нерв, обично видлива кај младите и може да биде асоцирана со периферна невропатија и срцеви аритмии. LHON има мајчински модел на наследување. Согласно со клиничките студии е утврдено дека повеќе заболуваат мажите отколку жените од ова заболување. Барем 19 различни митохондријални ДНК мутации се најдени кај LHON, од кои 5 се есенцијални и водат до болест. Интересно е да се спомене дека овие 5 мутации се асоцирани со различни степени на клиничко оштетување. Сите сродници на мајката заболени од LHON немаат загуба на видот и сите индивидуи со иста митохондријална мутација немаат ист клинички тек. Во клиничката слика се јавува акутна или субакутна болка, загуба на централниот вид со централен скотом. На офталмолошки преглед се забележува перипапиларна телеангиектазија, микроангиопатија и псевдоедем.
Терапија на митохондријалните заболувања
Додека генската терапија се усовршува, развојот на куративната терапија на митохондријалните заболувања треба да ја достигне во блиска иднина. Денешното постапување со овие метаболички нарушувања е насочено кон добивање оптимум енергетска ефикасност од дисфункцијата на митохондриите. Од другите мерки пациентот мора да превенира појава на треска, исцрпувачки вежби и лекови што го инхибираат митохондријалниот метаболизам. Рестрикциите во диетата се повеќе корисни кај заболувањата на липидниот метаболизам, како што се: оксидацијата на масните киселини или дефектите во карнитинскиот циклус. Се препорачуваат диети ослободени од масни киселини со долги вериги.
Кај нарушувањата на респираторната верига коензимот Q има широка употребата, иако во една двојно слепа плацебо-контролирана студија резултатите од употреба на коензимот Q се покажаа контрадикторни.
Во одделни случаи витамините K3, B2, C и E се покажале како корисни во терапијата. Егзогеното давање на карнитин може драматично да ги подобри симптомите кај пациентите со примарен или со секундарен карнитински дефицит. Давањето на дихлорацетат има средно поволен ефект кај случаите со Leigh-ова болест (9).
Генетичко советување
Бидејќи митохондријалните заболувања често водат до фенотипски промени и често се наследни, се јавува потреба од генетичко советување на афецираните семејства. Емпириските ризици неодамна беа предвидени за MELAS, MERRF синдромите и Leber-овата оптичка невропатија. Кај MELAS и MERRF високите нивоа на мутирана мт ДНК во крвта на мајките е асоцирана со зголемена фреквенција на афецирани потомци. Хроничната прогресивна надворешна офталмоплегија и Kearns-Sayre-овиот синдром главно претставуваат спорадични заболувања без зголемен ризик за репојавност во потомството. Кај Leigh-овата болест дефинирањето на молекуларниот дефект е круцијално во генетичкото советување, поради тоа што оваа болест се пренесува мајчински, автосомно рецесивно и Х-врзано.
Пренаталната дијагноза била претставена кај два случаи на митохондријално заболување, но според мислењето на повеќе автори ова е под знак на прашање, заради несигурната корелација во односот помеѓу мутираната ДНК во хорионските ресички и во клинички релевантните ткива како на пример во мозокот (10, 11).
Заклучок
Врз основа на изнесениот литературен преглед за митохондријалните заболувања може да се заклучи дека:
- тоа не се ретки заболувања;
- имаат големо епидемиолошко значење во дефектологијата;
- тие се последица на мутација на митохондријалната ДНК или т.н. 25 хромозом;
- најчести клинички манифестации се миопатиите, енцефалопатиите и кардиомиопатиите;
- успехот во терапијата лежи во иднината на генската терапија.
ЛИТЕРАТУРА
- Guyton AC. Medicinska fiziologija. Medicinska knjiga, Beograd-Zagreb, 1989;24-25
- Sack GH. Medical genetics. Mc Graw-Hill, 1999; 159-166
- Zergollern L i sur. Medicinska genetika 2. Školska knjiga, Zagreb, 1994; 37-38
- Лакоски А. Психогенетика. Скопје, 1998; 71-73
- Lopez de Munian A. Classification of mitochondrial diseases. Rev Neurol 1998; 26 Suppl 1: 9-14
- Howell N. Human mitochondrial diseases: answering questions and questioning answers. Int Rev Cytol 1999; 186: 49-116
- Treem WR, Sokol RJ. Disorders of the mitochondria. Semin Liver Dis 1998; 18 (3): 237-253
- Suomalainen A. Mitochondrial DNA and disease. Ann Med 1997; 29 (3): 235-246
- Munoz A, Bautista J. Treatment of mitochondrial disease. Rev Neurol 1998; Suppl 1: 87-91
- Klopstock T, Gasser T. Genetic counseling and prenatal diagnosis in mitochondrial disease. Nervenarzt 1999; 70 (6): 504-508
- White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, Di Mauro S, Dahl HH, Thorburn DR. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet 1999; 65 (2): 474-482
Follow Us
Share Us
Journal metrics
-
 SNIP 0.059
SNIP 0.059 -
IPP 0.07
-
 SJR 0.13
SJR 0.13 -
 h5-index 7
h5-index 7 -
 Google-based impact factor: 0.68
Google-based impact factor: 0.68
Indexed in





![]()
10 Most Read Articles
- PARENTAL ACCEPTANCE / REJECTION AND EMOTIONAL INTELLIGENCE AMONG ADOLESCENTS WITH AND WITHOUT DELINQUENT BEHAVIOR
- RELATIONSHIP BETWEEN LIFE BUILDING SKILLS AND SOCIAL ADJUSTMENT OF STUDENTS WITH HEARING IMPAIRMENT: IMPLICATIONS FOR COUNSELING
- EXPERIENCES FROM THE EDUCATIONAL SYSTEM – NARRATIVES OF PARENTS WITH CHILDREN WITH DISABILITIES IN CROATIA
- INOVATIONS IN THERAPY OF AUTISM
- DIAGNOSTIC AND TREATMENT OPTIONS IN AUTISTIC SPECTRUM DISORDERS – AN OVERVIEW
- AUTISM AND TUBEROUS SCLEROSIS
- THE DURATION AND PHASES OF QUALITATIVE RESEARCH
- REVIEW OF SPECIAL EDUCATION PROGRAMS IN JORDAN: CURRENT PRACTICES, CHALLENGES, AND PROSPECTS
- REHABILITATION OF PERSONS WITH CEREBRAL PALSY
- DISORDERED ATTENTION AS NEUROPSYCHOLOGICAL COGNITIVE DISFUNCTION